Factsheet about variant Creutzfeldt-Jakob disease
Variant Creutzfeldt-Jakob disease (vCJD) is a relatively new and rare neurological disease, classified as a Transmissible Spongiform Encephalopathy (TSE). It was first identified in March 1996 in the UK, when 10 cases of a new disease with neurological symptoms were reported and soon associated with the Bovine Spongiform Encephalopathy (BSE), “mad cow”-disease.
Causative agents of vCJD are prions, composed of misfolded prion proteins (PrPSc), which form aggregates in neurological tissue leading to progressive brain damage and characteristic signs and symptoms of the disease. Prions are stable and relatively resistant to proteases, high temperatures, UV radiation, and commonly used disinfectants.
Patients with vCJD have prominent psychiatric (frequently depression, anxiety and withdrawal) or sensory symptoms and delayed onset of neurologic abnormalities, including ataxia within weeks or months, and dementia and myoclonus late in the illness. The disease always progresses to death. Disease duration is 14 months on average. vCJD tends to affect younger individuals, with an average age of onset of around 28 years, compared to sporadic CJD, which tends to affect middle-aged and elderly individuals.
The definite diagnosis of vCJD requires post-mortem examination of brain tissue.
The incubation period for vCJD after food borne exposure is thought to be around 10 years. No vaccine or treatment is available.
Most reported vCJD cases appear to have been infected through the consumption of bovine meat products contaminated with the agent of BSE. In three cases, reported by the UK, the mode of transmission is thought to be through receipt of blood from an asymptomatic, infected donor.
Factsheet
The Infective agent
Bovine Spongiform Encephalopathy (BSE), variant CJD and transmissible spongiform encephalopathies in general are prion diseases, caused by aggregates of misfolded prion protein.
First hypothesized by Prusiner in 1982, as the cause of scrapie [1], prions are unconventional transmissible infectious agents composed largely or exclusively of misfolded prion protein (PrPSc), which produces the disease by propagating the abnormal conformation to the normal cellular prion protein (PrPC). The conversion from normal prion protein to abnormal prion protein form proceeds as a chain-reaction and leads to a higher content of specific forms of protein (beta sheet forms). Aggregates of PrPSc proteins are responsible for brain damage leading to the characteristic signs and symptoms of the disease [2].
Forms of human prion disease include sporadic, variant, inherited and iatrogenic (transmitted through certain medical procedures) CJD. Experimental evidence indicates that the prion strain responsible for the BSE epizootic in cows is also responsible for the vCJD cases in humans [3] and is different from the causative agent in other TSEs and sporadic CJD.
Transmission and exposure
Most reported vCJD cases appear to have been infected through the consumption of cattle products contaminated with the agent of BSE [4]. In three cases, reported in the UK, the mode of transmission is thought to have been through receipt of a blood transfusion derived from an asymptomatic, but infected donor [5-8] .
Variant CJD has been transmitted experimentally to different animal species, including wild type mice, transgenic mice and non-human primates [9].
During infection with prion diseases, infectious titres of prion protein are present in peripheral tissues (particularly lymphoid organs and spleen) before a progressive rise in brain titres finally results in clinical disease. Subclinical or carrier states may have major public health implications for public health, particularly regarding potential iatrogenic transmission from apparently healthy persons [10].
Epidemiology
Since 1996 and as of August 2013, a total of 229 cases of variant CJD cases have been identified from 11 countries: 177 from the United Kingdom, 27 from France, 4 from Ireland, 4 from the United States, 5 from Spain, 3 in the Netherlands, 2 each from Portugal, Italy and Canada, and 1 each from Japan, Taiwan and Saudi Arabia.
Table 1 - Worldwide total number of cases, as of January 2015
|
Country |
Total number primary cases (Number alive) |
Total number secondary cases: blood transfusion (Number alive) | Cumulative residence in UK 6 months during period 1980-1996 |
|---|---|---|---|
| UK | 174 (0) | 3 (0) | 177 |
| France | 26(0) | - |
1 |
| Republic of Ireland |
4(0) |
- | 2 |
| Italy | 2 (0) | - | 0 |
| USA | 4 † (0) | - | 2 |
|
Canada |
2 (0) |
- |
1 |
| Saudi Arabia | 1 (0) | - | 0 |
| Japan | 1* (0) | - | 0 |
| Netherlands | 3 (0) | - | 0 |
| Portugal | 2 (0) | - | 0 |
| Spain | 5 (0) | - | 0 |
| Taiwan | 1 (0) | - | 1 |
| TOTAL | 226 (0) | 3 (0) | 184 |
† the third US patient with vCJD was born and raised in Saudi Arabia and has lived permanently in the United States since late 2005. According to the US case-report, the patient was most likely infected as a child when living in Saudi Arabia.
In the fourth US patient the history indicated that exposure to infection most likely occurred prior to moving to the USA.*the case from Japan had resided in the UK for 24 days in the period 1980-1996.
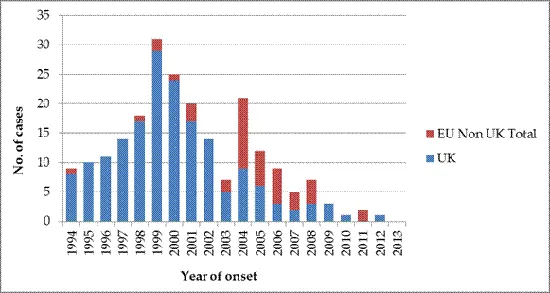
In 1999 the epidemic of vCJD peaked in the UK, declining progressively with only one probable incident case in 2012, diagnosed and notified in 2013 (as of 1 January 2015). There is a clear delay between the UK epidemic and the occurrence of cases in other EU countries, which peaked in 2004, with 11 incident cases. The most affected country, besides UK, was France with a total of 27 cases.
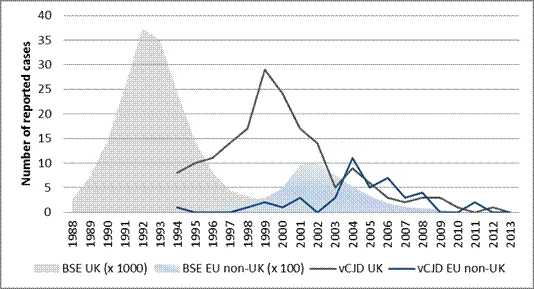
Similarly the trend of BSE cases shows a peak of reported cases in 1992 in UK, followed by a lower peak in 2002 in the other EU countries (EU non-UK) [11]. The time period between peaks of BSE and vCJD cases in the UK is around eight years; while the time period between peaks of BSE and vCJD cases in EU non-UK countries is only two years.
In the EU vCJD has tended to affect younger individuals with median age of onset at 28 year (range 11-74). Sporadic CJD usually affects middle-aged and elderly individuals.
The median duration of illness in the EU is 14 month (range 6-114).
Clinical features and diagnosis
Incubation period: The estimated mean incubation period (defined as the time from infection to death) for infection via primary transmission is estimated to be approximately the delay from the peak of exposure to BSE which occurred in 1989/1990 to the peak in the vCJD deaths which occurred in 2000 [12-14]..The incubation period in transfusion transmitted vCJD has been between 6.5 and 8 years.
In a recent study of French vCJD cases, the incubation period has been estimated to be around 13 years (95% CI: 9,7-17,9 years) [15]. The incubation period is not dependant on age [16], however age-dependent susceptibility/exposure is estimated to be highest in teenagers and young adults in line with previous estimates [13].
Clinical presentation: Clinical descriptions of all forms of CJD have been developed by the National Creutzfeldt-Jakob Disease Surveillance Unit in the UK: patients with vCJD have prominent early psychiatric (depression, anxiety and withdrawal) or sensory symptoms with a delayed onset of neurological abnormalities. Ataxia develops at around 6 months, and dementia and myoclonus are seen later in the illness. The disease always progresses to death [17,18].
Diagnosis: Clinical and investigative features, which are included in the diagnosis criteria, may be indicative of the diagnosis of vCJD but are never definitive as case confirmation requires neuropathological examination (link to case definition). Important diagnostic tools include:
- EEG shows a typical periodic pattern in many cases of sporadic CJD, but this is rarely seen in vCJD.
- MRI brain scan shows pulvinar high signal in 90% of cases of vCJD and basal ganglia high signal in about 70% of cases of sporadic CJD.
- CSF14-3-3 protein is elevated in 90% of cases of sporadic CJD and 50% of vCJD cases.
Neuropathological examination, usually after autopsy, is necessary for diagnostic confirmation and is also the definitive method for distinguishing between sporadic and variant CJD.
Treatment: Only palliative treatment is possible. No curative treatment is available.
Preventive measures
Since 1989, several control and prevention measures have been implemented in the EU [19]. The feed ban is the basic preventive measure laid down against TSE and consists of a ban on the use of processed animal protein (PAP) in feed for farmed animals. Based on scientific findings that linked the spread of BSE to the consumption of contaminated PAP feed, a ban on the feeding of mammalian processed animal protein to cattle, sheep and goats was introduced in the UK in 1988 and in other member states in July 1994. Aiming at eradication of certain TSE, the ban was expanded in January 2001 with the feeding of all processed animal proteins to all farmed animals being prohibited, with certain limited exceptions (TSE regulation). In 2009 a further ban on the use of milk and milk products coming from classical scrapie infected flocks for feeding ruminants was set out [20,21]. As a result, the number of BSE affected cattle in the UK has declined steadily, with only 3 reported cases so far in 2013 (figure 3) [11]. The probability of food-borne exposure to prion diseases in the EU appears now to be very limited.
To date, there has been no known association between primary vCJD and occupation, medicines, immunising agents, gelatin, or surgery (including the use of catgut sutures). Nevertheless, vCJD infection has been observed in three recipients of blood transfusions from two donors who later developed the disease and one blood recipient who died of another cause without clinical symptoms, but who at autopsy had prions in spleen and lymph nodes [5-8,22] . The possibility of a risk has been assessed for plasma products, human organ and tissue transplants and contaminated surgical instruments or devices, but, to date, no definite transmission through these routes has been identified. However, prions are remarkably stable and relatively resistant to proteases, high temperatures and UV-radiation, as well as to commonly used disinfectants [4].
Key areas of uncertainty
The vCJD epidemic peaked in the EU in the period 1999-2004. The incidence has decreased steadily after the implementation of stringent prevention measures and the epidemic is now in its tail [12]. There are, however, remaining concerns and uncertainties:
- Genetic susceptibility: Up to now, all tested cases of vCJD have been methionine homozygotes at codon 129 of the PrP gene (a genotype present in approximately 40% of Caucasian populations). Recently, a possible vCJD case was reported in a 30-year old man who was heterozygous at codon 129 [23]. However, the case was not confirmed. If other genotypes are not completely resistant to infection but have longer incubation periods, as suggested in kuru and growth hormone-related CJD, subsequent epidemics in these genotypes may yet appear [12].
- Sub-clinical forms: Persons with latent disease undergoing surgical procedures could result in instrument contamination with a risk of secondary transmission [10,24] . The first evidence of transmission through blood transfusion raised concerns about iatrogenic transmission and there is the possibility of pre-clinical or sub-clinical prion-associated infection in an unknown proportion of the population [4].
- Under-ascertainment of cases: While ascertainment of cases is probably good for young adults, it is still possible that cases of vCJD in old people are missed, because of the small proportion of those dying from dementia who undergo post mortem neuropathological examination [10].
- Clinical features: It is possible that infection with BSE prions could produce symptoms different than those described until now for vCJD, for example in persons with genotypes other than MM at codon 129 of the prion protein gene [10,24].
During recent years, evidence has been accumulating that protein misfolding is central in the causation of a range of neurodegenerative disorders, including Alzheimer’s disease and Parkinson’s disease.
- Partial lift of feed bans: In 2008 and 2009 the TSE regulation was amended to provide for a derogation from the general prohibition of feeding animal protein to ruminants with a view to allowing the feeding of proteins derived from fish to young animals of ruminant species, subject to certain conditions [20]. This change in policy may increase the risk of TSE transmission to cattle. Due to the long latency of the vCJD the effects of any potential human exposure to contaminated animal product will be evident in the years to come.
- Atypical BSE: As part of the massive testing for BSE at slaughterhouses in the EU, abnormal prion positive cows without symptoms have been detected in Europe and North America. These prion strains were initially detected in Italy and referred as to bovine amyloidotic spongiform encephalopathy (BASE). Animal tests in mice have shown an evolution of BASE strain towards BSE strain and it has been suggested that the BASE strain is the origin of the BSE strain that has caused the epidemics in humans and animals [25] . A recent study suggests a potential link between BASE and a subset of sporadic CJD cases based on the biochemical similarity of the strains [26] .
EU Surveillance for variant CJD
The surveillance of vCJD became mandatory in the EU in 2000 (Commission Decision 2000/96/EC). The EuroCJD network, established in 1993, was evaluated and transferred under the responsibility of ECDC in 2007.
The surveillance is currently performed by the EuroCJD network, co-ordinated from the National CJD Research & Surveillance Unit based in Edinburgh and funded by ECDC. There are now 29 collaborating centres from EU Member States and EEA/EFTA countries. The case definitions for vCJD apply to all EU countries (link).
The reporting, including all historical cases was established in TESSy in 2012.
References
1. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982 04/09;216(4542):136-44.
2. Castilla J, Gonzalez-Romero D, Saa P, Morales R, De Castro J, Soto C. Crossing the species barrier by prp(sc) replication in vitro generates unique infectious prions. Cell. 2008 Sep 5;134(5):757-68. PubMed PMID: 18775309. Pubmed Central PMCID: PMC2740631. Epub 2008/09/09. eng.
3. Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, et al. Transmissions to mice indicate that 'new variant' cjd is caused by the bse agent. Nature. 1997 10/02;389(6650):498-501.
4. Collee JG, Bradley R, Liberski PP. Variant cjd (vcjd) and bovine spongiform encephalopathy (bse): 10 and 20 years on: Part 2. Folia neuropathologica / Association of Polish Neuropathologists and Medical Research Centre, Polish Academy of Sciences. 2006;44(2):102-10. PubMed PMID: 16823692. Epub 2006/07/11. eng.
5. Llewelyn CA, Hewitt PE, Knight RSG, Amar K, Cousens S, Mackenzie J, et al. Possible transmission of variant creutzfeldt-jakob disease by blood transfusion. Lancet. 2004 02/07;363(9407):417-21.
6. Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vcjd after blood transfusion in a prnp codon 129 heterozygous patient. Lancet. 2004 08/07;364(9433):527-9.
7. Chohan G, Llewelyn C, Mackenzie J, Cousens S, Kennedy A, Will R, et al. Variant creutzfeldt-jakob disease in a transfusion recipient: Coincidence or cause? Transfusion. 2010 05/;50(5):1003-6.
8. Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Creutzfeldt-jakob disease and blood transfusion: Results of the uk transfusion medicine epidemiological review study. Vox Sang. 2006 Oct;91(3):221-30. PubMed PMID: 16958834. Epub 2006/09/09. eng.
9. Heymann DLAPHA. Control of communicable diseases manual. Washington, DC: American Public Health Association; 2004.
10. Smith PG, Cousens SN, d' Huillard Aignaux JN, Ward HJ, Will RG. The epidemiology of variant creutzfeldt-jakob disease. Curr Top Microbiol Immunol. 2004;284:161-91. PubMed PMID: 15148992. Epub 2004/05/20. eng.
11. Annual reports of member states on bse and scrapie 2013.
12. Garske T, Ghani AC. Uncertainty in the tail of the variant creutzfeldt-jakob disease epidemic in the uk. PloS one. 2010;5(12):e15626. PubMed PMID: 21203419. Pubmed Central PMCID: PMC3009744. Epub 2011/01/05. eng.
13. Ghani AC, Ferguson NM, Donnelly CA, Anderson RM. Short-term projections for variant creutzfeldt-jakob disease onsets. Statistical methods in medical research. 2003 Jun;12(3):191-201. PubMed PMID: 12828241. Epub 2003/06/28. eng.
14. Valleron AJ, Boelle PY, Will R, Cesbron JY. Estimation of epidemic size and incubation time based on age characteristics of vcjd in the united kingdom. Science. 2001 Nov 23;294(5547):1726-8. PubMed PMID: 11721058. Epub 2001/11/27. eng.
15. Chadeau-Hyam M, Clarke PS, Guihenneu-Jouyaux C, Cousens SN, Will RG, Ghani AC. An application of hidden markov models to the french variant creutzfeldt-jakob disease epidemic. Appl Statist. 2010 Jan 2010;59(Part 5):15. eng.
16. Ghani AC, Ferguson NM, Donnelly CA, Anderson RM. Predicted vcjd mortality in Great Britain. Nature. 2000 Aug 10;406(6796):583-4. PubMed PMID: 10949288. Epub 2000/08/19. eng.
17. Ironside JW. Variant creutzfeldt-jakob disease: Risk of transmission by blood transfusion and blood therapies. Haemophilia : the official journal of the World Federation of Hemophilia. 2006 Mar;12 Suppl 1:8-15; discussion 26-8. PubMed PMID: 16445812. Epub 2006/02/01. eng.
18. Turner MLAAoBB. Creutzfeldt-jakob disease : Managing the risk of transmission by blood, plasma, and tissues. Bethesda, Md.: AABB Press; 2006.
19. Bradley R, Collee JG, Liberski PP. Variant cjd (vcjd) and bovine spongiform encephalopathy (bse): 10 and 20 years on: Part 1. Folia neuropathologica / Association of Polish Neuropathologists and Medical Research Centre, Polish Academy of Sciences. 2006;44(2):93-101. PubMed PMID: 16823691. Epub 2006/07/11. eng.
20. Chronological legislation eu about bse 2013, EC.
21. Commission decision 2000/418/ec regulating the use of material presenting risks as regards transmissible spongiform encephalopathies, 2000/418/EC OJ(2000).
22. Soldan K. Uk blood donors identified as at increased risk of vcjd are to be notified of their status 2005 [updated 07/; cited 10 7].
23. Kaski D Fau - Mead S, Mead S Fau - Hyare H, Hyare H Fau - Cooper S, Cooper S Fau - Jampana R, Jampana R Fau - Overell J, Overell J Fau - Knight R, et al. Variant cjd in an individual heterozygous for prnp codon 129. (1474-547X (Electronic)).
24. Hilton DA. Pathogenesis and prevalence of variant creutzfeldt-jakob disease. The Journal of pathology. 2006 Jan;208(2):134-41. PubMed PMID: 16362983. Epub 2005/12/20. eng.
25. Capobianco R, Casalone C, Suardi S, Mangieri M, Miccolo C, Limido L, et al. Conversion of the base prion strain into the bse strain: The origin of bse? PLoS pathogens. 2007 Mar;3(3):e31. PubMed PMID: 17352534. Pubmed Central PMCID: PMC1817656. Epub 2007/03/14. eng.
26. Comoy EE, Casalone C, Lescoutra-Etchegaray N, Zanusso G, Freire S, Marce D, et al. Atypical bse (base) transmitted from asymptomatic aging cattle to a primate. PloS one. 2008;3(8):e3017. PubMed PMID: 18714385. Pubmed Central PMCID: PMC2515088. Epub 2008/08/21. eng.